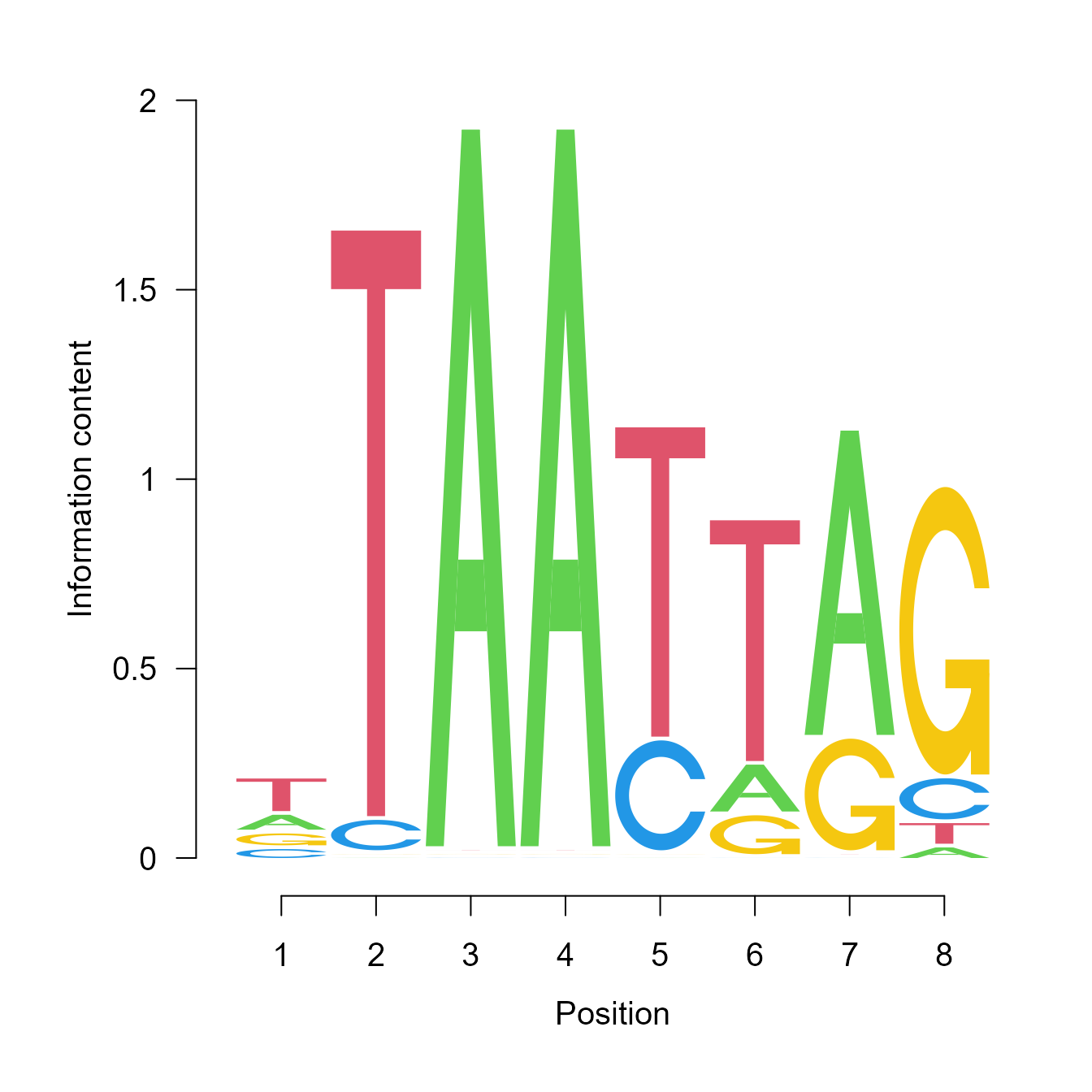Monogenic MOTIF analysis
Motif.Rmd1.Create Linkage Object
The chromatin accessibility data is a normalized bulk ATAC-seq count matrix, which a prior count of 5 is added to the raw counts, then put into a “counts per million”, then log2 transformed, then quantile normalized. The gene expression data is a normalized bulk RNA-seq count matrix, which is log2(fpkm+1) transformed. Two tab-delimited text/csv input files (chromatin accessibility matrix and gene expression matrix) are required before running Linkage. The gene expression matrix file is a tab-delimited multi-column data matrix, which the first column represents gene symbols and the following columns represent normalized or raw expression levels of genes for each sample. The chromatin accessibility matrix file is a tab-delimited multi-column data matrix as well, which the first three columns represent chromosome name, start coordinate on the chromosome and end coordinate on the chromosome of the peaks respectively; the remaining columns of the chromatin accessibility matrix file represent normalized or raw chromatin accessibility levels of peaks for each sample. Each element of ready-to-analysis files will appear in Chromatin Accessibility Matrix panel and Gene Expression Matrix panel.
library(linkage)
library(LinkageData)
ATAC.seq <- BreastCancerATAC()
RNA.seq <- BreastCancerRNA()
LinkageObject <-
CreateLinkageObject(
ATAC_count = ATAC.seq,
RNA_count = RNA.seq,
Species = "Homo",
id_type = "ensembl_gene_id"
)
LinkageObject## An LinkageObject
## 59161 gene 215920 peak
## Active gene: 0
## Active peak: positive peak 0 negetive peak 0
## Active TF: 02.Regulatory Peaks Search
Regulatory Peaks Search allows users to detect all potential regulatory DNA regions for specific genes. When given an input gene and targeting DNA regions, Linkage will automatically perform canonical correlation analysis between each quantitative chromatin accessibility measure in the region and the quantitative expression level of the gene across all samples. Users can easily adjust the region of interest and customize the correlation algorithm (Spearman / Pearson / Kendall). Then, all the statistically significant results are listed in the Potential Cis-regulatory Regions panel. With clicking on a specific region of this panel, users can view the scatter plot of quantitative chromatin accessibility and gene expression from the Correlation Plot panel. The rho and FDR for correlation analysis also be shown on the scatter plot.
gene_list <- c("TSPAN6", "CD99", "KLHL13")
LinkageObject <-
RegulatoryPeak(
LinkageObject = LinkageObject,
gene_list = gene_list,
genelist_idtype = "external_gene_name"
)3.Motif Analysis Module
Motif Analysis Module supports users to visualize the enriched transcription factor motifs within potential regulatory peaks. With clicking on a specific peak, users can view the location and binding score information of each enriched transcription factor of this region. Once users select one of the transcription factors, the corresponding sequence logo of this cis-regulatory element will appear in the Sequence-logo Plot panel.
library(DT)
df <- SingleMotifAnalysis(LinkageObject@cor.peak$TSPAN6, "Homo")
brks1 <-
quantile(df$score,
probs = seq(.05, .95, .05),
na.rm = TRUE)
DT::datatable(
df,
selection = "single",
extensions = c("Scroller", "RowReorder"),
option = list(
rowReorder = TRUE,
deferRender = TRUE,
scrollY = 295,
scroller = TRUE,
scrollX = TRUE,
searchHighlight = TRUE,
orderClasses = TRUE,
autoWidth = F
)
) %>% formatStyle(
names(df)[8],
background = styleColorBar(range(brks1), 'lightblue'),
backgroundSize = '90% 100%',
backgroundRepeat = 'no-repeat',
backgroundPosition = 'center'
)
SeqLogoPlot("MA0618.1")