TCGA-BRCA guided tutorial
TCGA-BRCA-guided-tutorial.Rmd1.Setup the Linkage Object
For this tutorial, we will be analyzing the dataset of Breast Cancer (BRCA) freely available from TCGA. The dataset contains chromatin accessibility and gene expression data from 72 patients with BRCA, which can be load from LinkageData. You can also found the raw data from here.
We start by loading in the data. The BreastCancerATAC() and BreastCancerRNA() function can load and return the available chromatin accessibility and gene expression matrices from Linkage. The chromatin accessibility matrix is a normalized bulk ATAC-seq count matrix, which a prior count of 5 is added to the raw counts, then put into a "counts per million", then log2 transformed, then quantile normalized. The gene expression matrix is a normalized bulk RNA-seq count matrix, which is log2(fpkm+1) transformed.
We next use the chromatin accessibility and gene expression matrix to create a Linkage object. The object serves as a container that contains both data (like the count matrix) and analysis (like regulatory peaks, or active TFBS).
library(Linkage)
# library(LinkageData)
# ATAC_count <- BreastCancerATAC()
# RNA_count <- BreastCancerRNA()
LinkageObject <-
CreateLinkageObject(
ATAC_count = Small_ATAC,
RNA_count = Small_RNA,
Species = "Homo",
id_type = "ensembl_gene_id"
)
LinkageObject## An LinkageObject
## 48 gene 32 peak
## Active gene: 0
## Active peak: positive peak 0 negetive peak 0
## Active TF: 02.Regulatory Peaks Search
Linkage allows you to search all potential regulatory peaks of given genes within a specified region.
In the example below, we detect the potential regulatory peaks of three different genes (“TSPAN6”, “CD99”, “KLHL13”) within a range of 500000 bp around them. Then we plot the top 6 significant results (sort by FDR).
gene_list <- c("TSPAN6", "CD99", "KLHL13")
LinkageObject <-
RegulatoryPeak(
LinkageObject = LinkageObject,
gene_list = gene_list,
genelist_idtype = "external_gene_name",
)
CorrPlot(LinkageObject, gene = "CD99")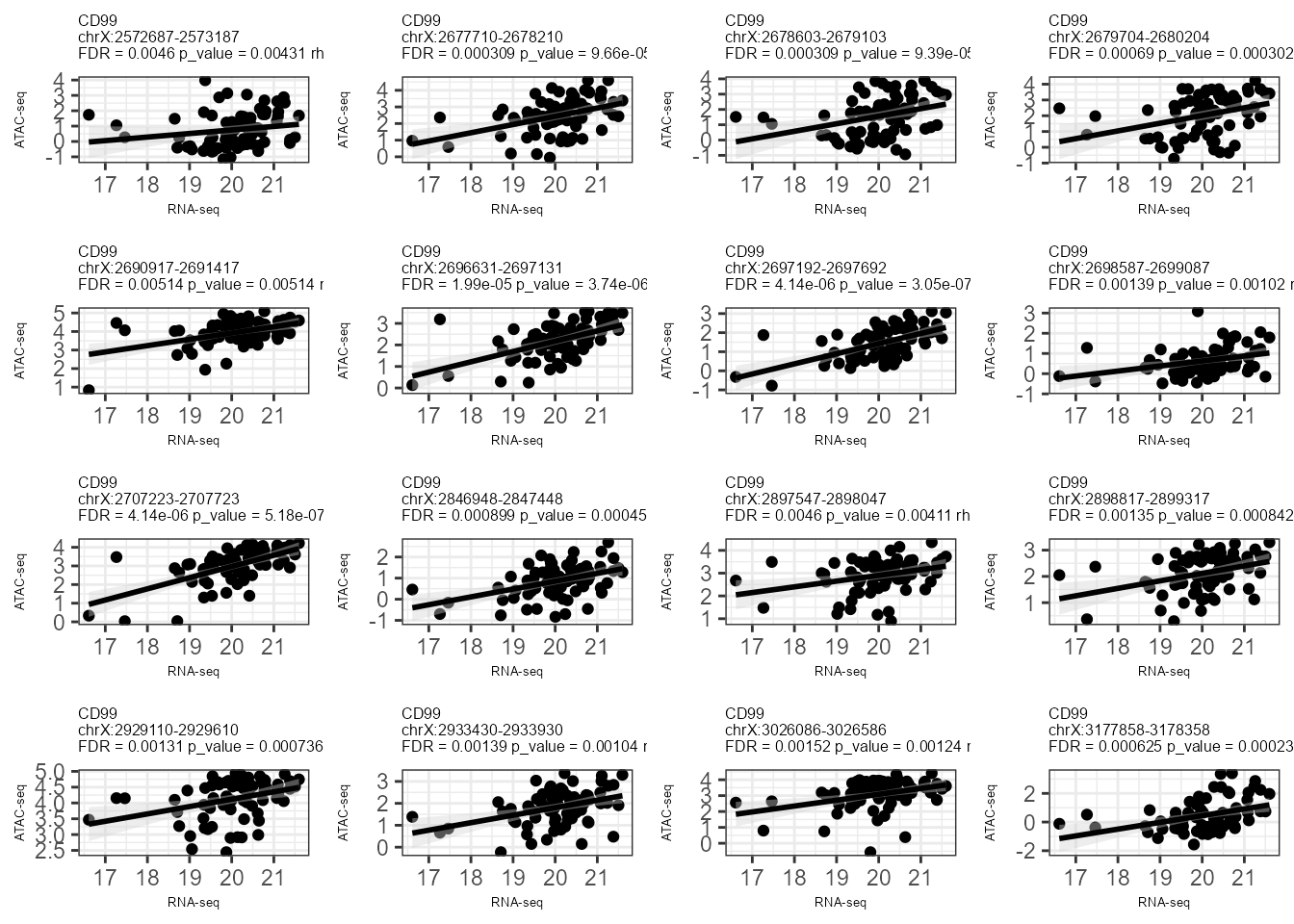
3.Regulatory Peaks Visualization
Now we can plot the Tn5 integration frequency and gene expression
level across regions of the genome using the TrackPlot()
function. For easier visualization, Linkage categorizes samples into
five groups based on the quantitative chromatin accessibility of the
specific regulatory peak from low to high. For each group, pseudo-bulk
accessibility tracks and expression boxplot of the target gene can be
viewed. Alongside these accessibility tracks we can visualize other
important information including gene annotation and peak
coordinates.
TrackPlot(
LinkageObject,
Geneid = "TSPAN6",
peakid = "chrX:100635908-100636408",
Species = "Homo"
)
4.Regulatory Peaks Annotation
Linkage can help you visualize the annotation of the predicational
regulatory peaks. The PeakAnnotation() perform the
annotation of all predicational regulatory peaks in terms of genomic
location features, which includes whether the peak is in the TSS, Exon,
5' UTR, 3' UTR, Intronic or Intergenic and the position and strand
information of the nearest gene of the peaks. You can produce the
upsetplot for visualizing the overlaps and distribution in annotation
for peaks by the AnnoUpsetPlot() function.
LinkageObject <- PeakAnnotation(LinkageObject, Species = "Homo")
AnnoUpsetPlot(LinkageObject = LinkageObject)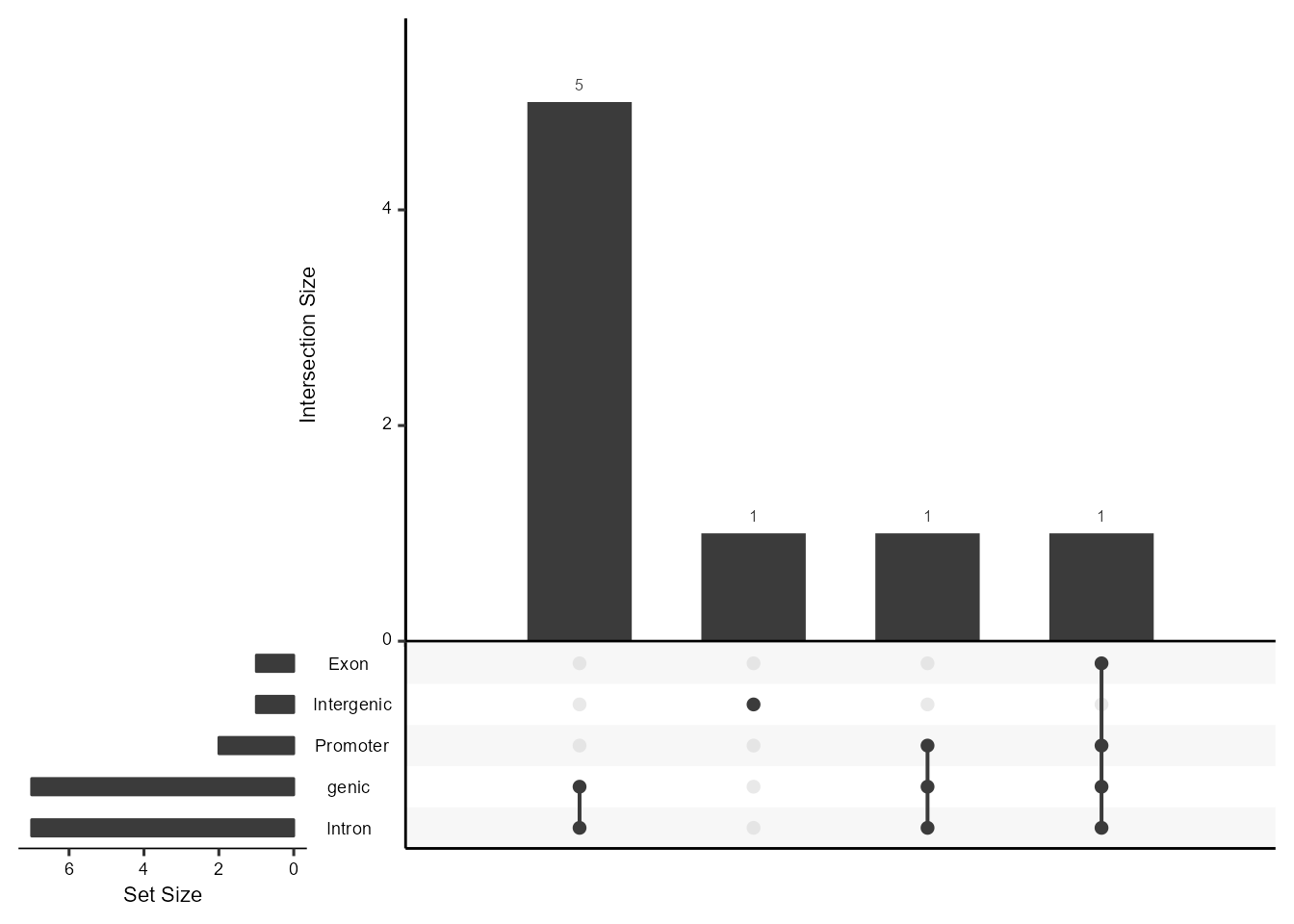
5.Motif Encichment Analysis
Next, you can visualize the enriched CREs within potential regulatory
peaks by the MotifEnrichment() function. CREs and the TFs
that binding on them play a central role in gene transcription
regulation, which can be detected by ATAC-seq data. This function can
help you view the location and binding score information of each
enriched TFBS of the given DNA region.
library(DT)
df <- MotifEnrichment(LinkageObject@cor.peak$TSPAN6, "Homo")
brks1 <-
quantile(df$score,
probs = seq(.05, .95, .05),
na.rm = TRUE)
DT::datatable(
df,
selection = "single",
extensions = c("Scroller", "RowReorder"),
option = list(
rowReorder = TRUE,
deferRender = TRUE,
scrollY = 295,
scroller = TRUE,
scrollX = TRUE,
searchHighlight = TRUE,
orderClasses = TRUE,
autoWidth = F
)
) %>% formatStyle(
names(df)[8],
background = styleColorBar(range(brks1), 'lightblue'),
backgroundSize = '90% 100%',
backgroundRepeat = 'no-repeat',
backgroundPosition = 'center'
)The SeqLogoPlot() function further help you to get the
sequence logo of the given transcription factors.
SeqLogoPlot("MA0618.1")